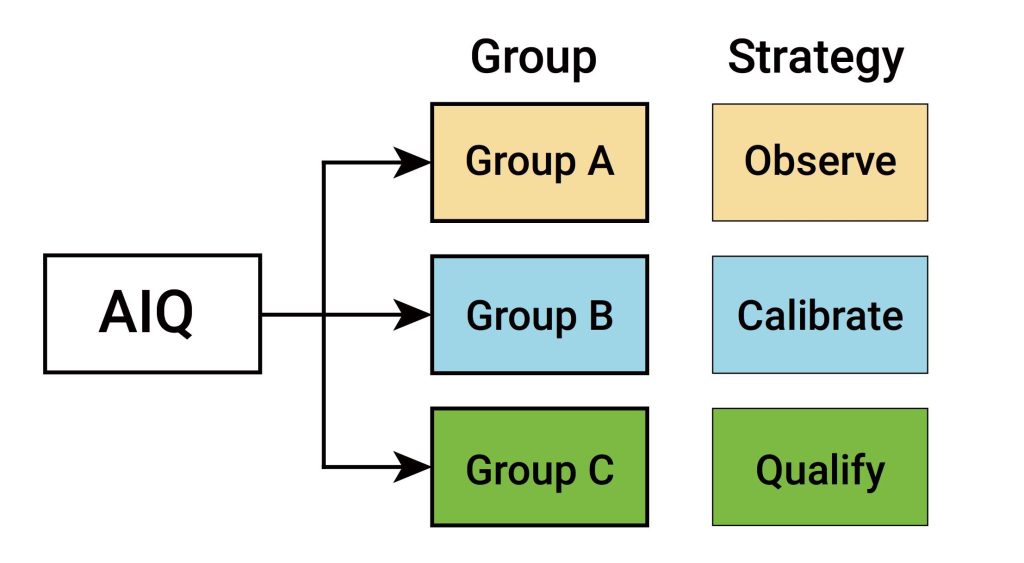
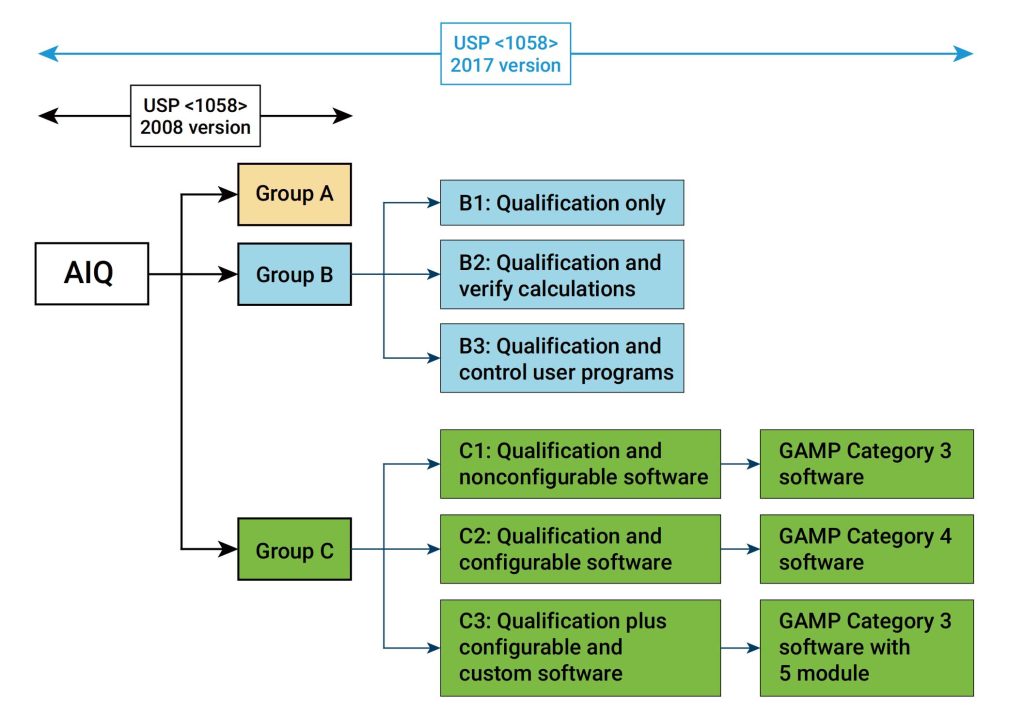
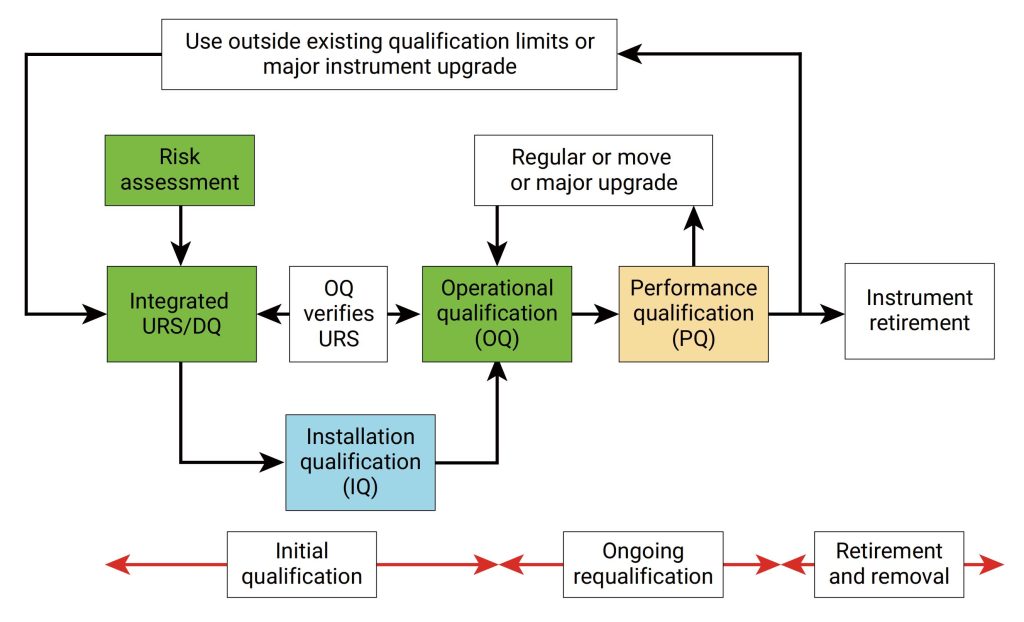
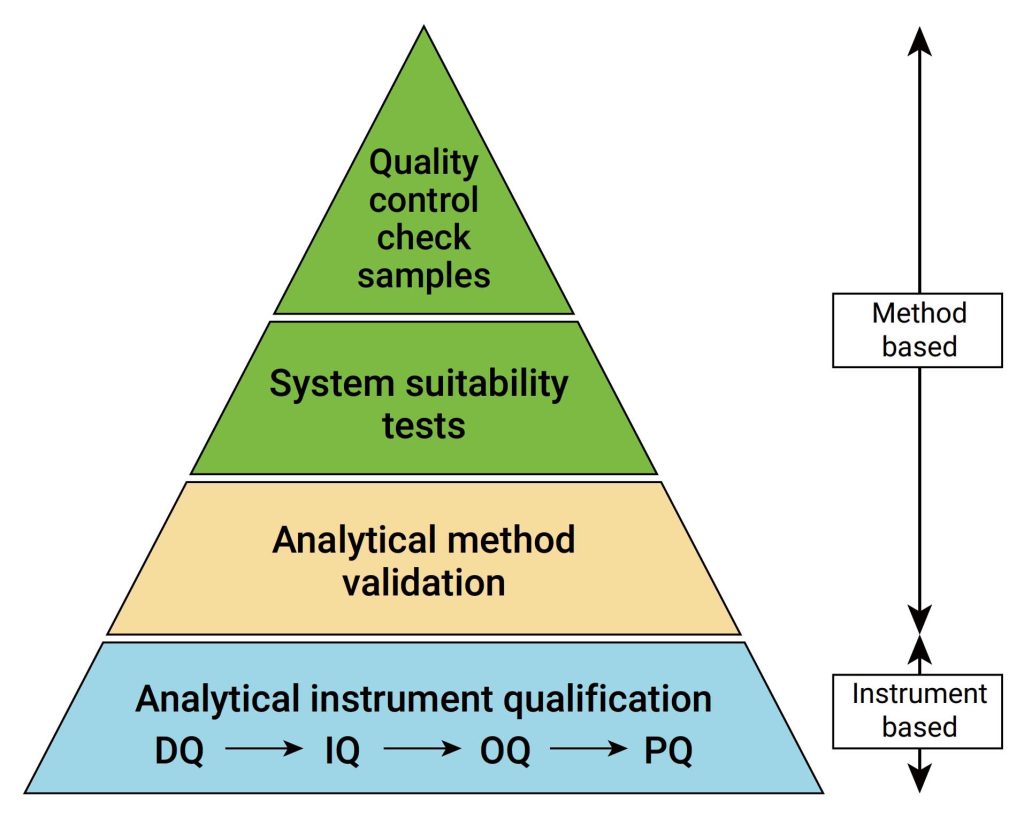
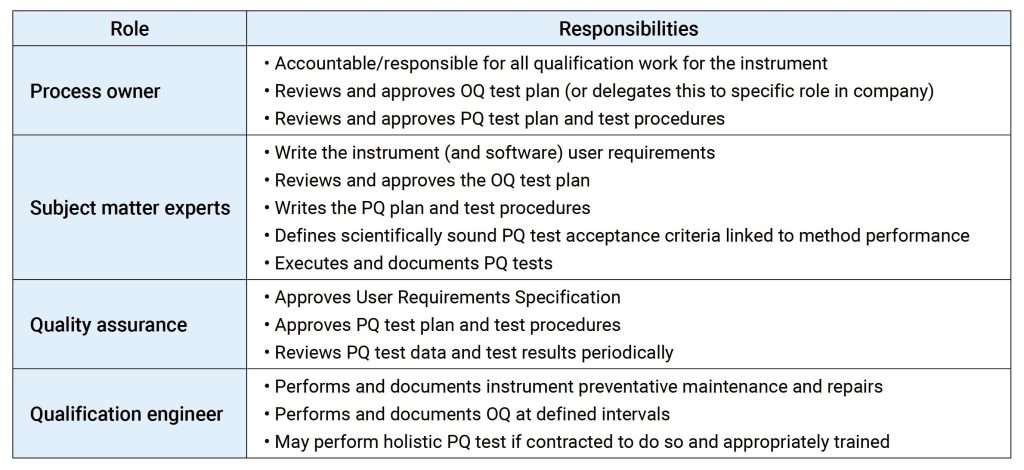
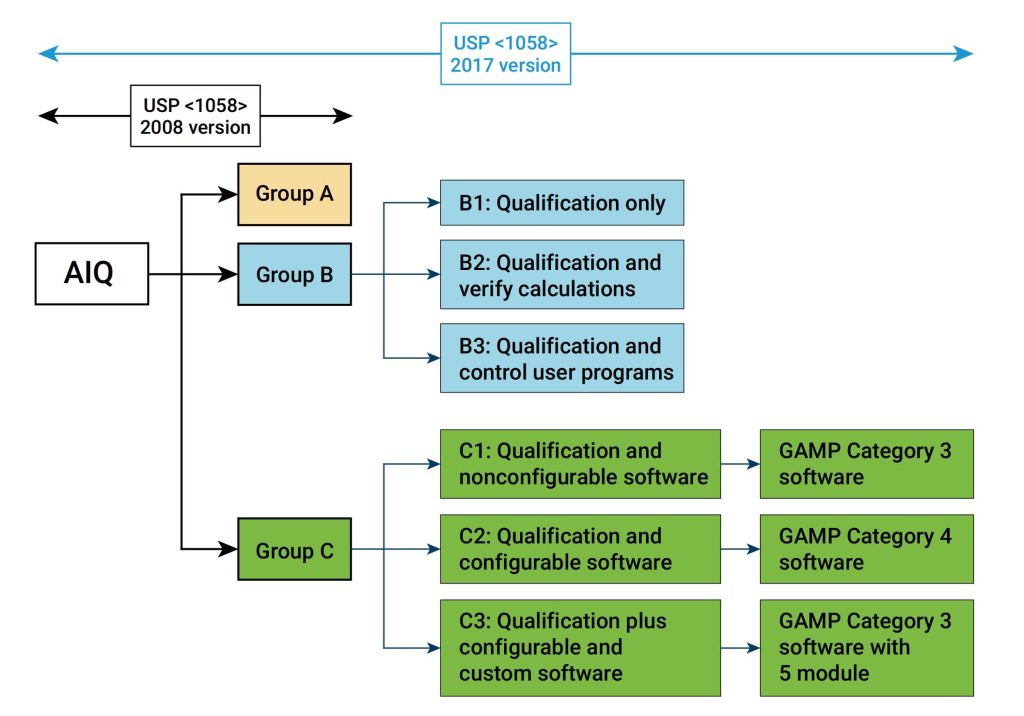
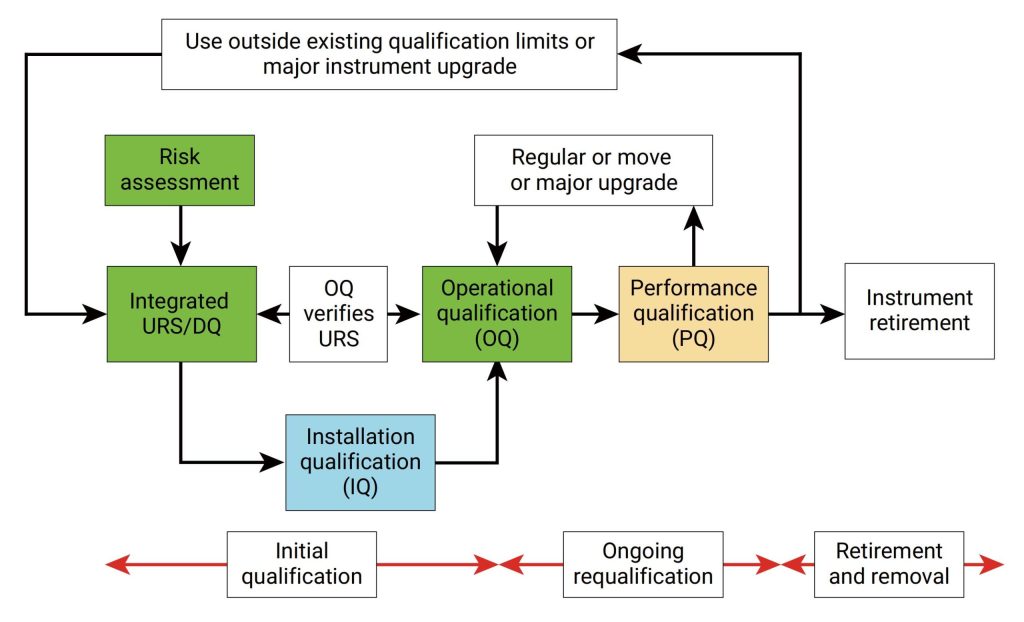
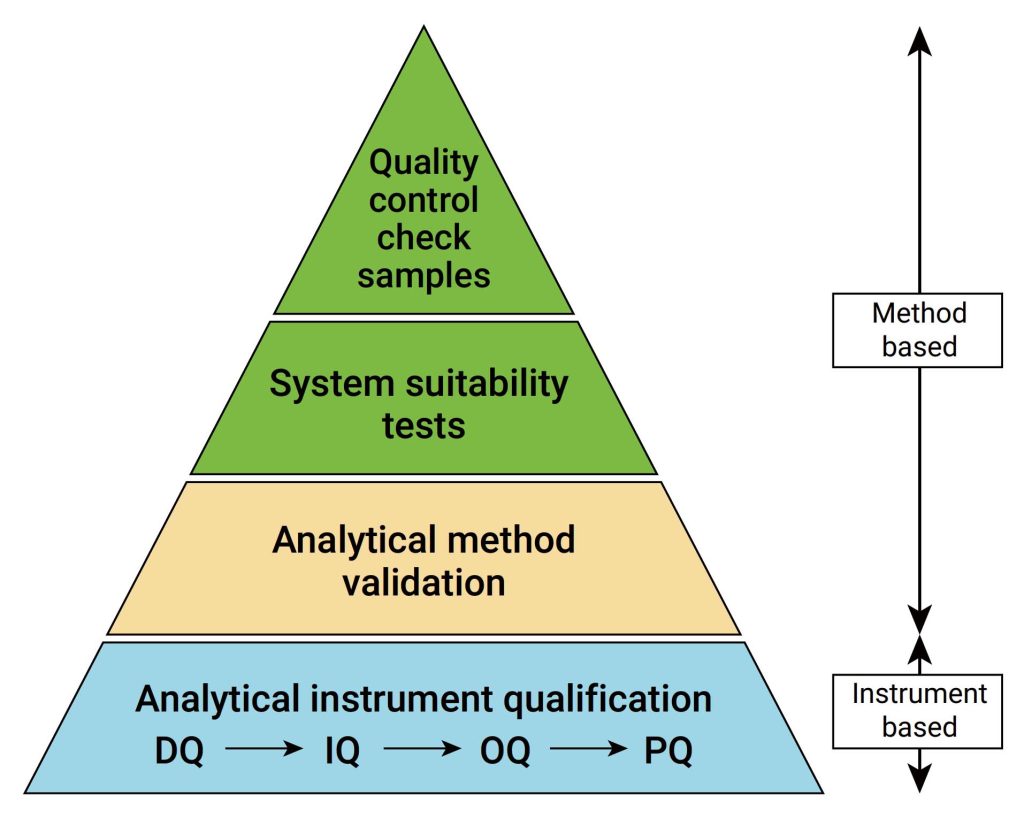
IQ是活动记录集,这些活动是确定交付的仪器符合设计和规格要求,并正确安装于所选择的环境中,且该环境是仪器所必需的,用户负责确保 IQ 已充分执行,并涵盖诸如仪器适用位置等项目。
IQ 将包含以下项目:,交货单和货物状况、现场安装要求、环境要求、服务和工具,组件和安装、软件安装、网络和数据存储,安装验证、其他文档中指定的信息,IQ适用于全新的或旧仪器,对于之前未进行确认或按现行行业标准确认不合格的任何现有仪器,应整理现有文档并进行风险评估,以确定最佳应对方案。
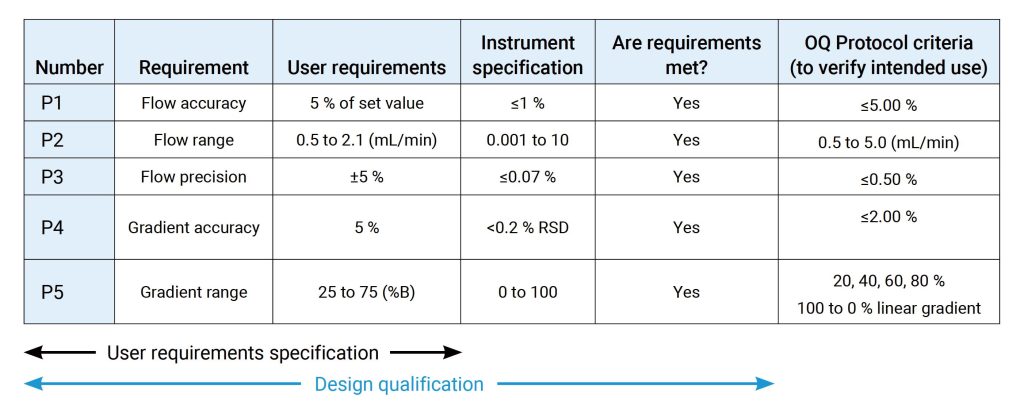
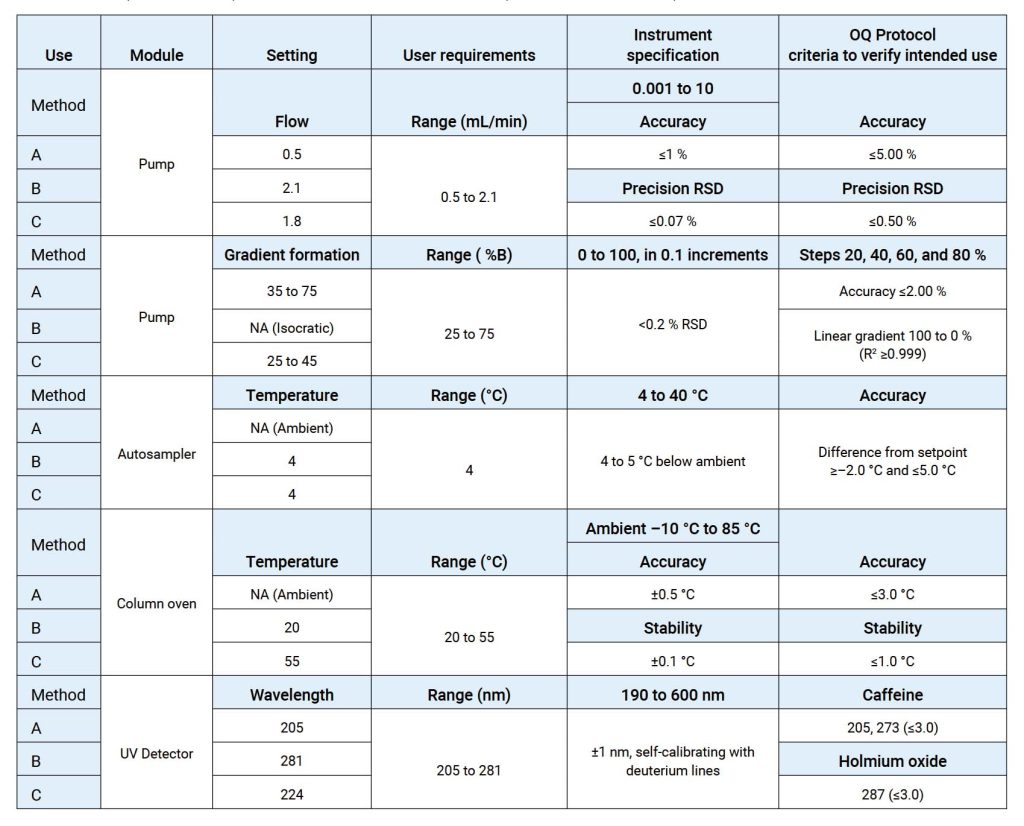
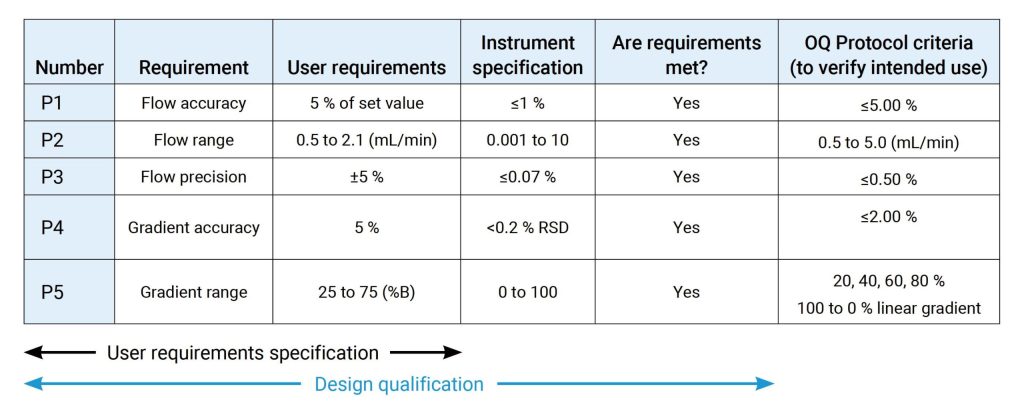
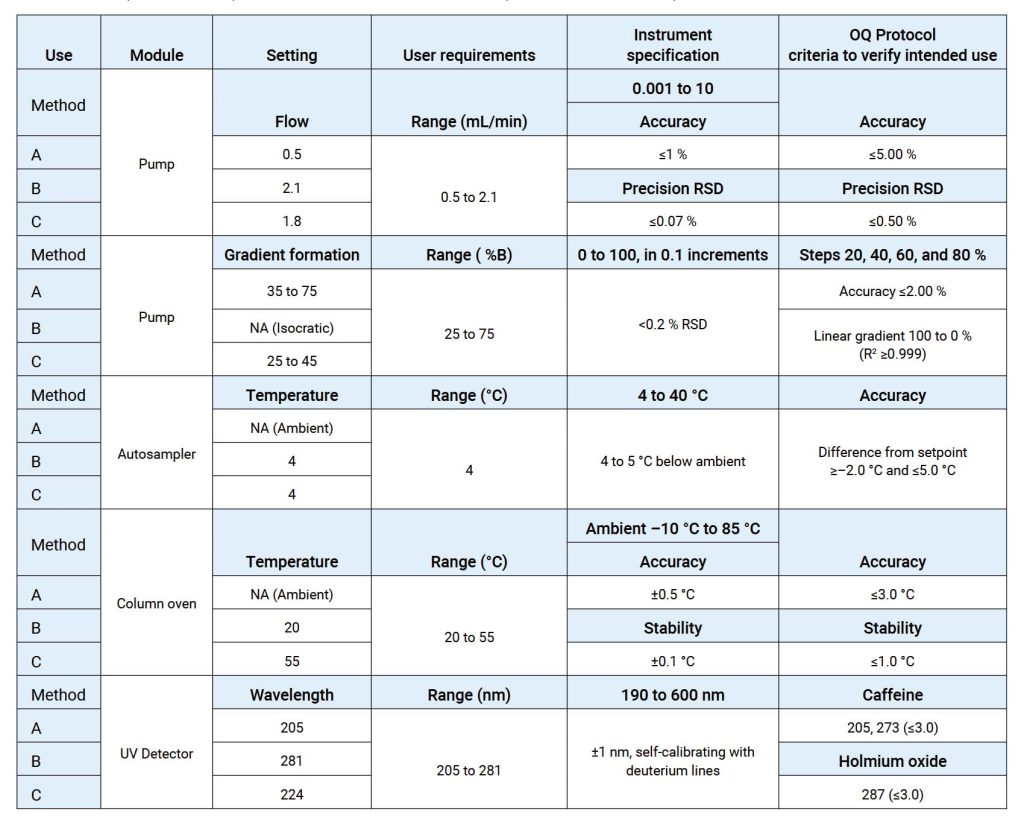
OQ是证明仪器可以在选定环境下,按照操作规程测试正常工作的活动记录集,OQ应证明对所选应用的适用性且体现URS,包括固定参数,这些测量仪器的不变参数,例如长度高度等,安全数据存储、备份和归档,根据书面程序在用户现场测试数据处理,例如存储、备份、审计跟踪和归档,仪器功能测试应测试用户要求的仪器功能,以验证仪器是否按照制造商的预期运行,OQ 测试可以是模块化的或整体的,仪器软件的任何配置都应在 OQ 之前进行并记录在案。
IQ是活动记录集,这些活动是确定交付的仪器符合设计和规格要求,并正确安装于所选择的环境中,且该环境是仪器所必需的,用户负责确保 IQ 已充分执行,并涵盖诸如仪器适用位置等项目。
IQ 将包含以下项目:,交货单和货物状况、现场安装要求、环境要求、服务和工具,组件和安装、软件安装、网络和数据存储,安装验证、其他文档中指定的信息,IQ适用于全新的或旧仪器,对于之前未进行确认或按现行行业标准确认不合格的任何现有仪器,应整理现有文档并进行风险评估,以确定最佳应对方案。
OQ是证明仪器可以在选定环境下,按照操作规程测试正常工作的活动记录集,OQ应证明对所选应用的适用性且体现URS,包括固定参数,这些测量仪器的不变参数,例如长度高度等,安全数据存储、备份和归档,根据书面程序在用户现场测试数据处理,例如存储、备份、审计跟踪和归档,仪器功能测试应测试用户要求的仪器功能,以验证仪器是否按照制造商的预期运行,OQ 测试可以是模块化的或整体的,仪器软件的任何配置都应在 OQ 之前进行并记录在案。