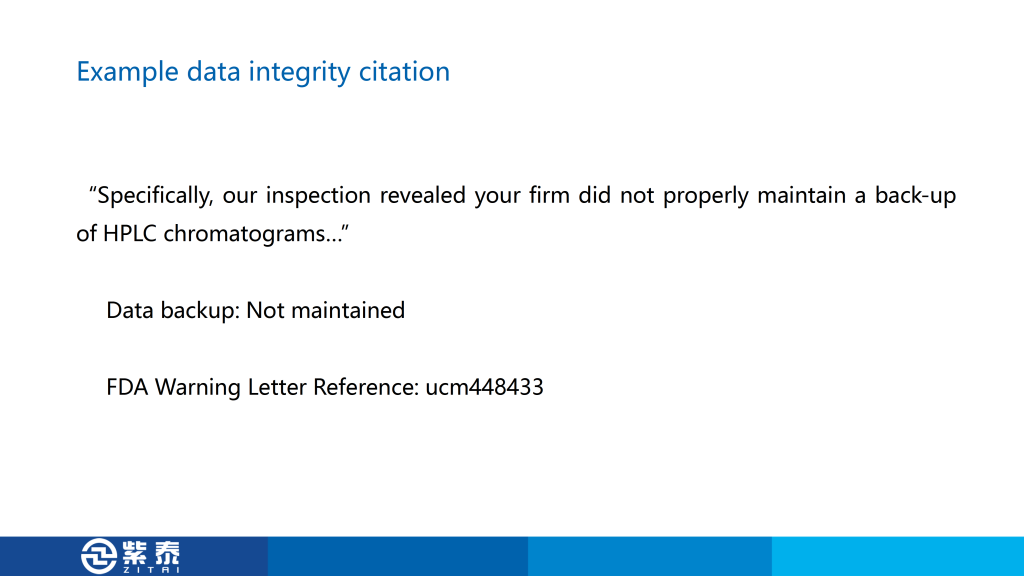
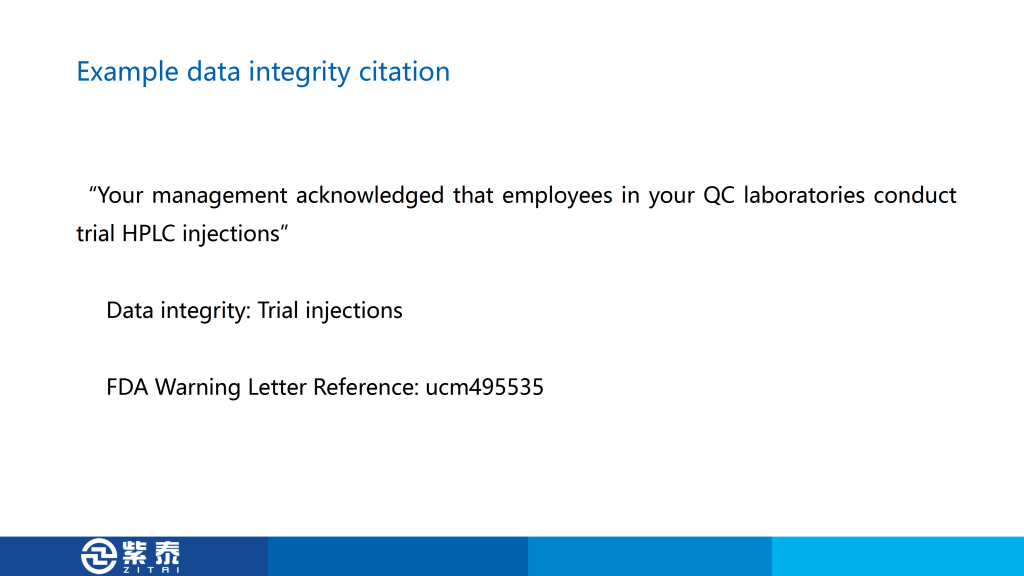
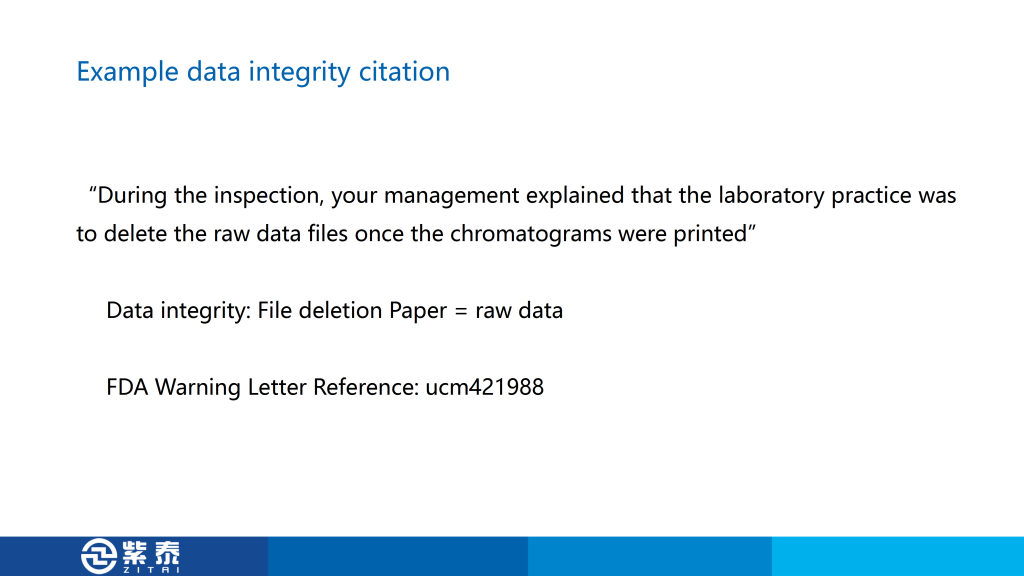
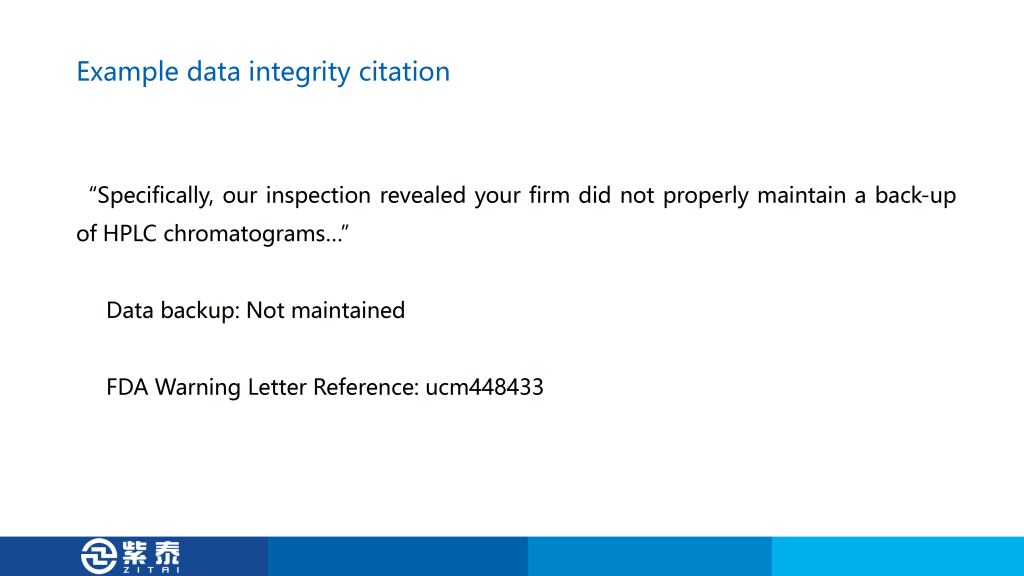
首先是第一个,在62种仪器的列表中,只有四种完全合格,另外五台仪器只经历了 DQ、IQ 和 OQ 步骤,这是认证不完全,也是我在认证中常见的问题,毕竟找人做认证需要花钱,所以很多公司会只做一部分仪器的3Q,这样节省费用,来检查的时候只给专家看做完的那几台设备,这个警告信就是针对这种现象,您如果有设备没有做过3Q可能会遇到的风险。
首先是第一个,在62种仪器的列表中,只有四种完全合格,另外五台仪器只经历了 DQ、IQ 和 OQ 步骤,这是认证不完全,也是我在认证中常见的问题,毕竟找人做认证需要花钱,所以很多公司会只做一部分仪器的3Q,这样节省费用,来检查的时候只给专家看做完的那几台设备,这个警告信就是针对这种现象,您如果有设备没有做过3Q可能会遇到的风险。