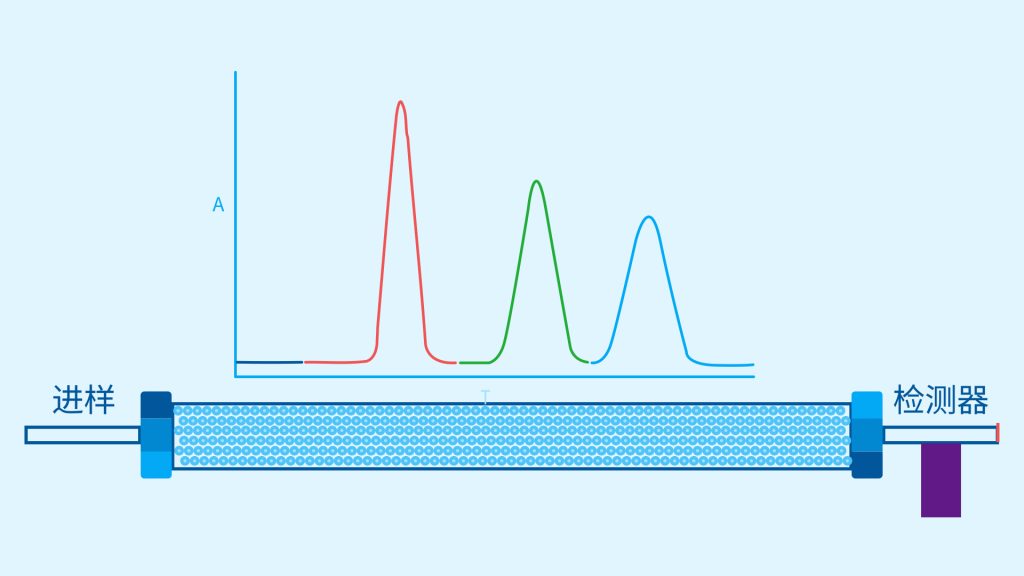
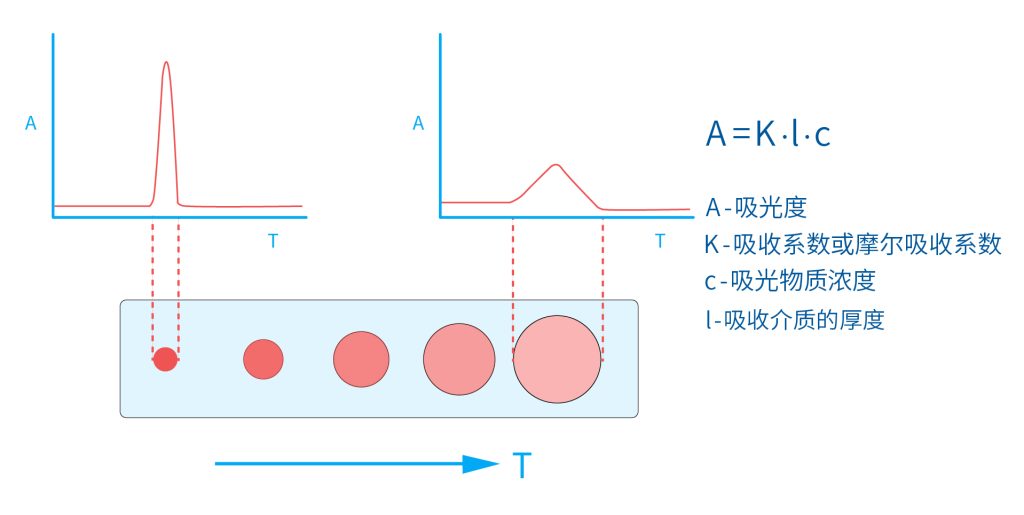
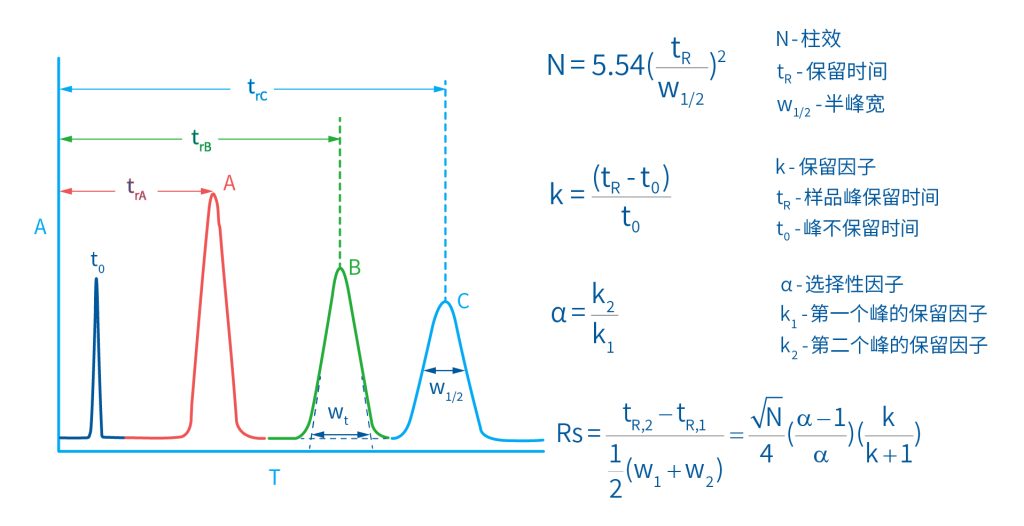
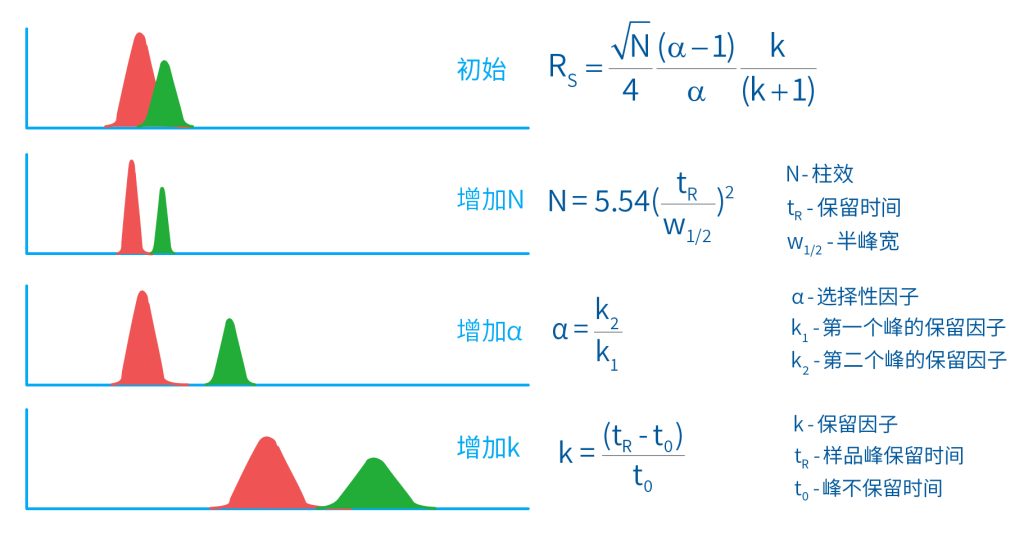
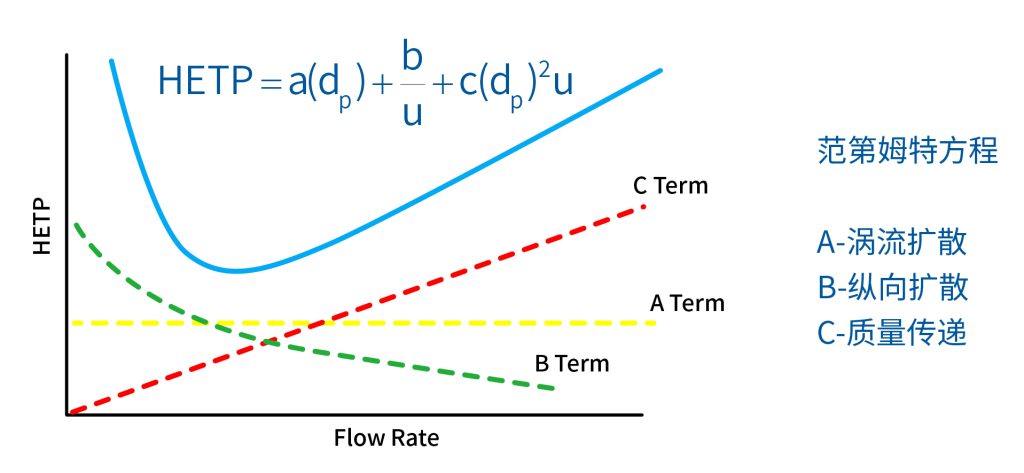
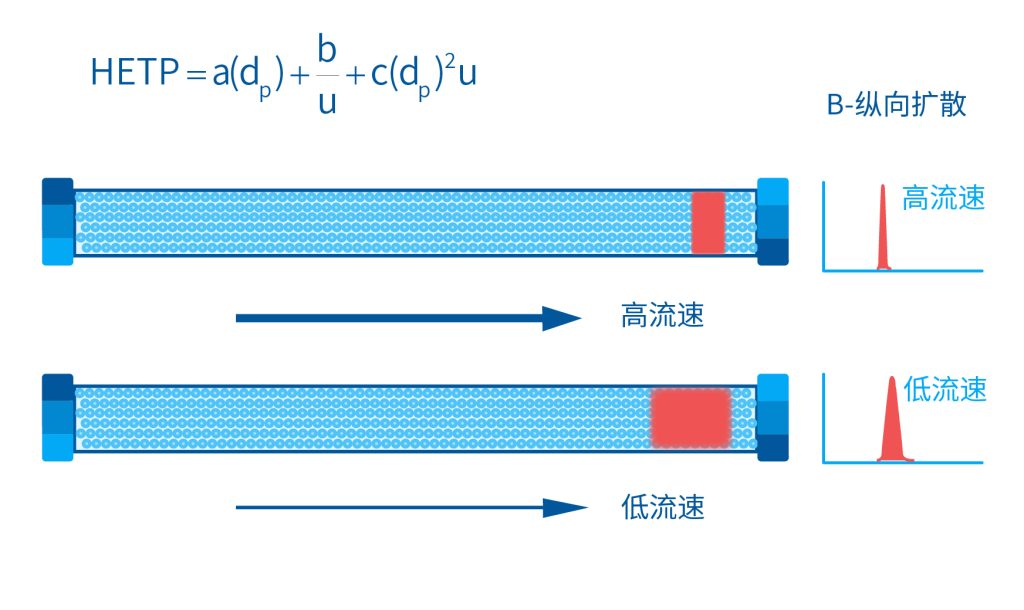
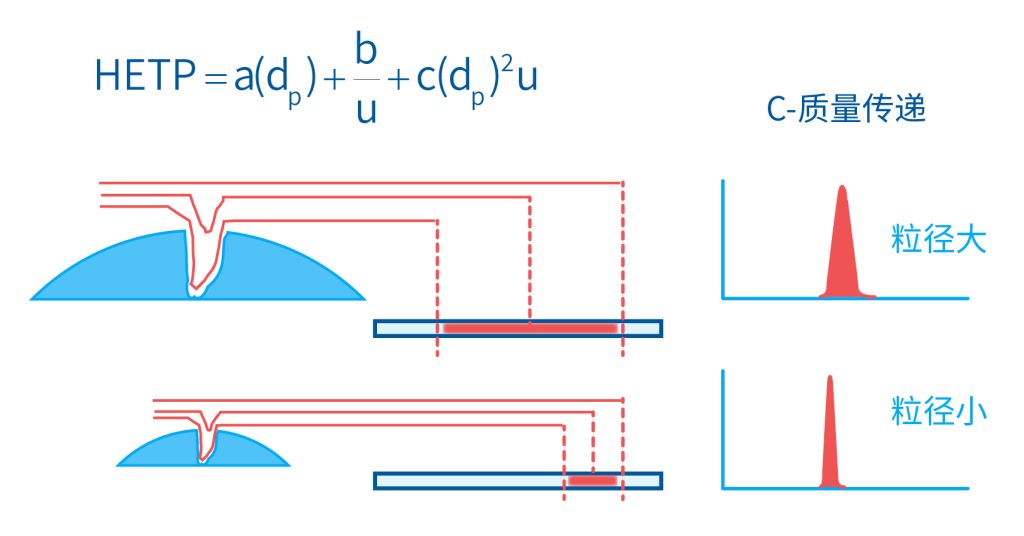
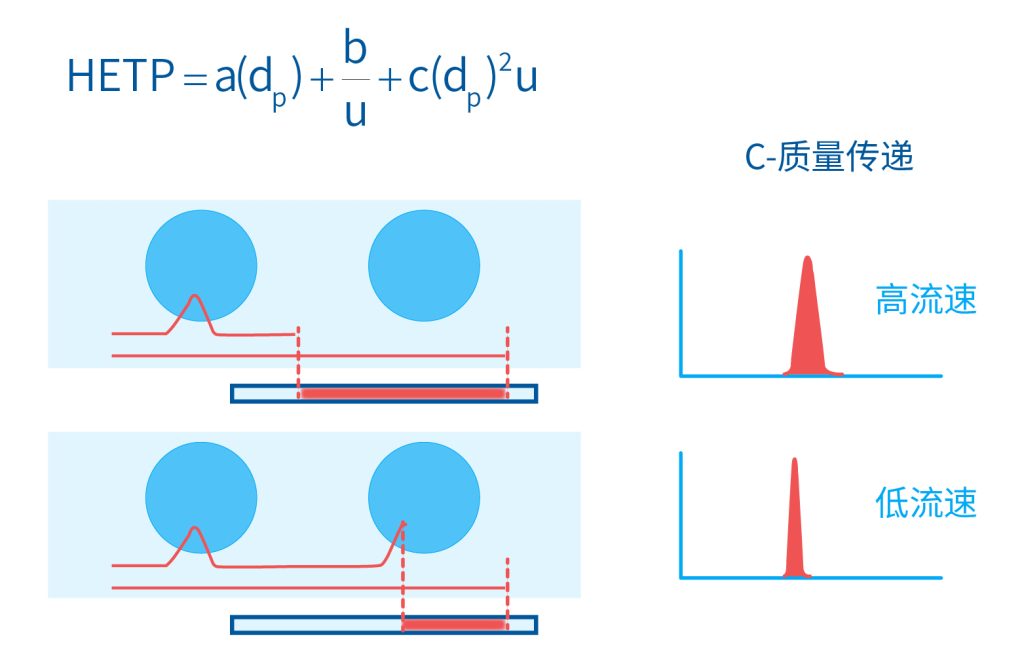
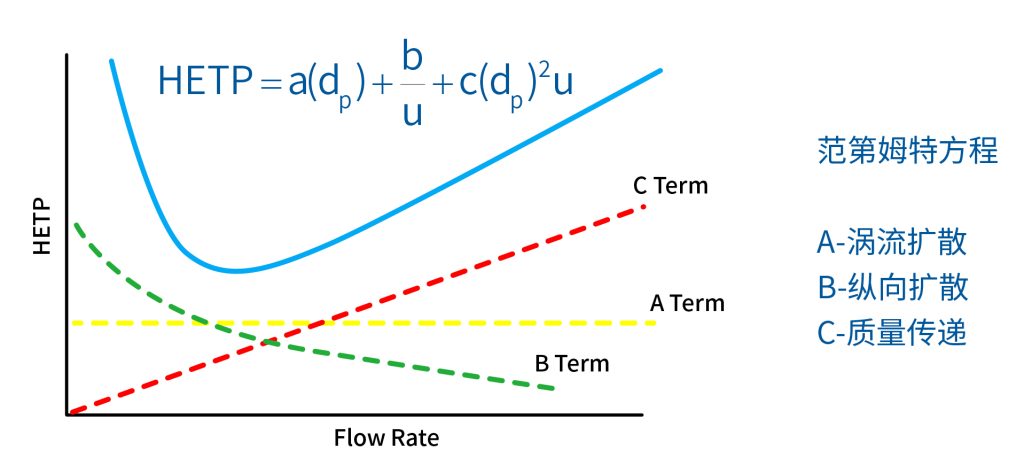
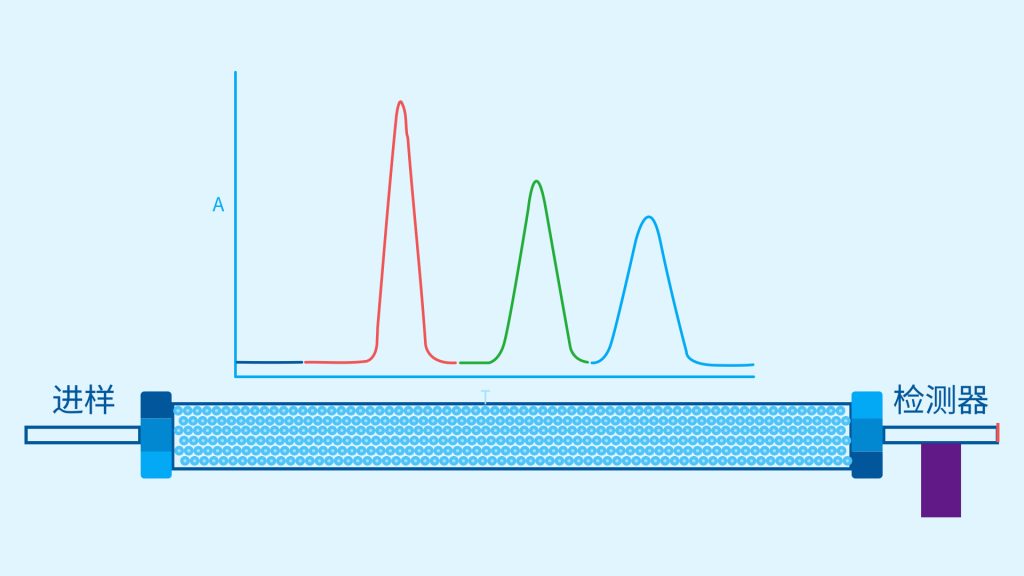
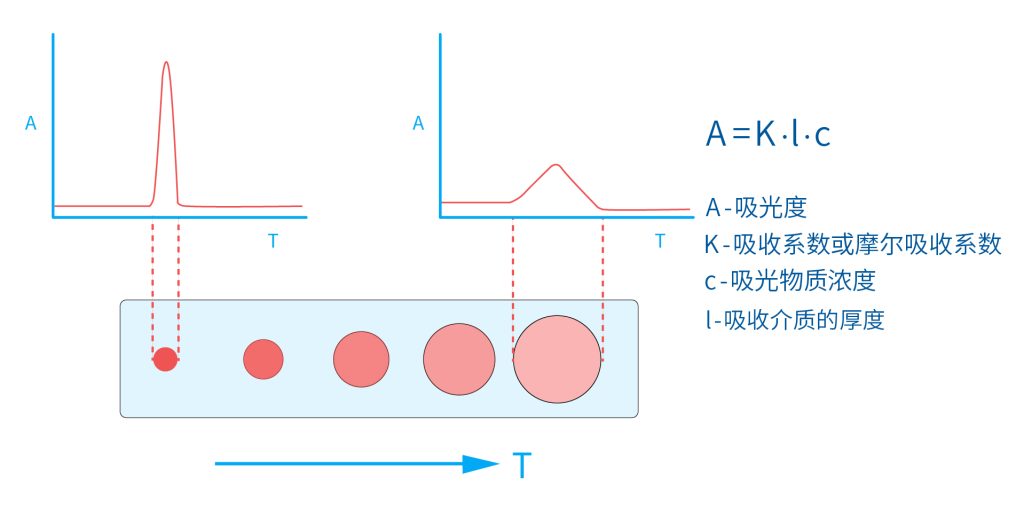
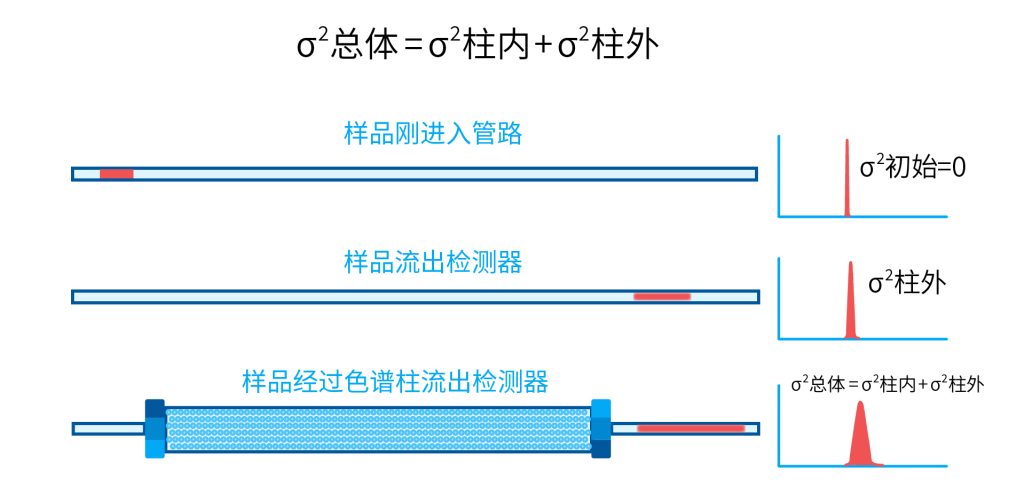
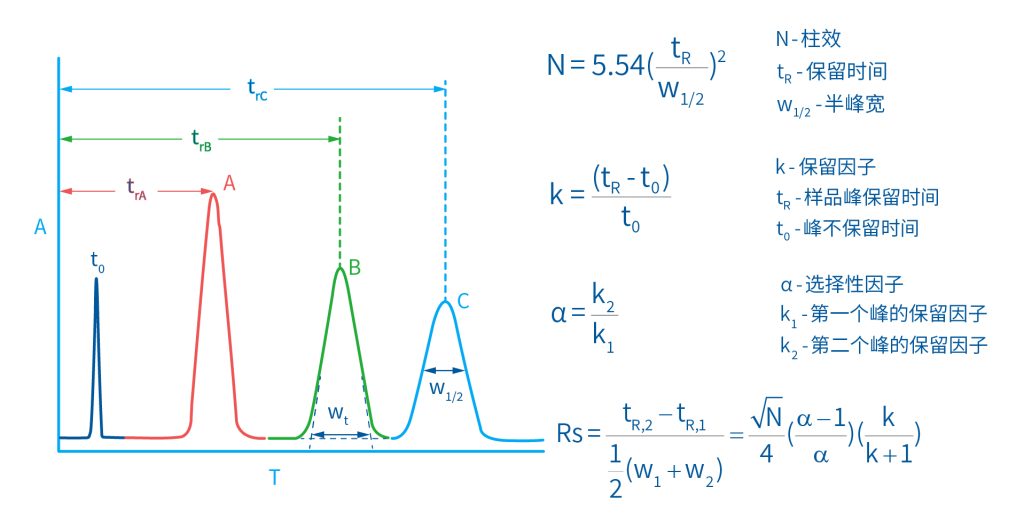
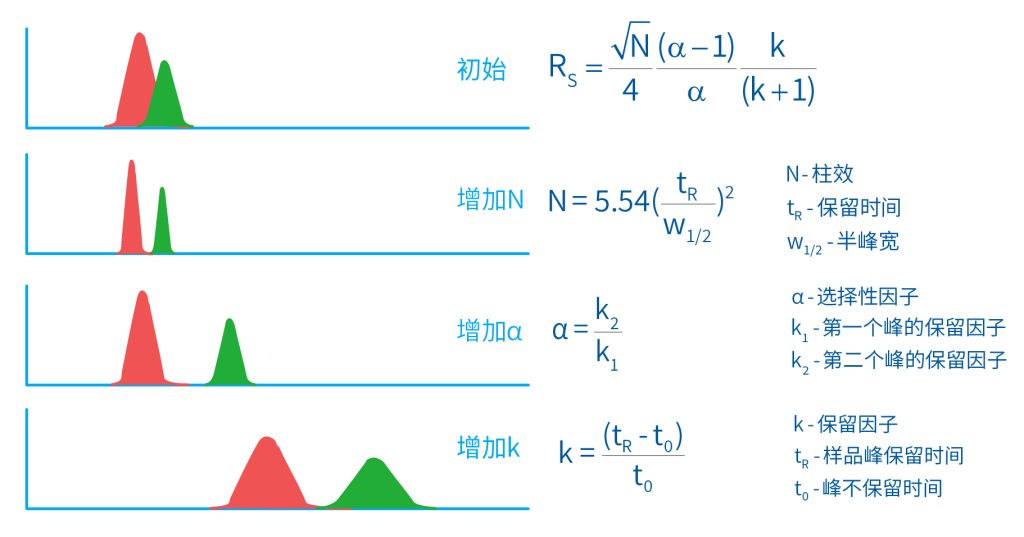
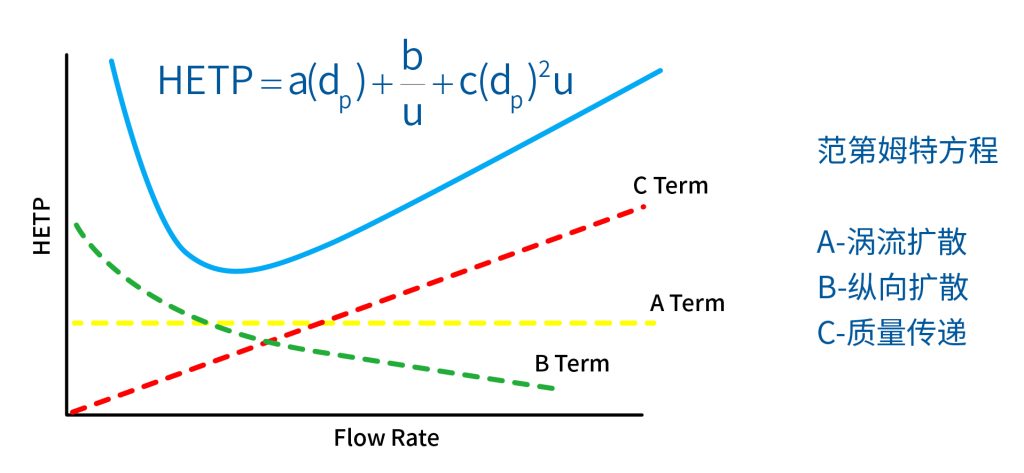
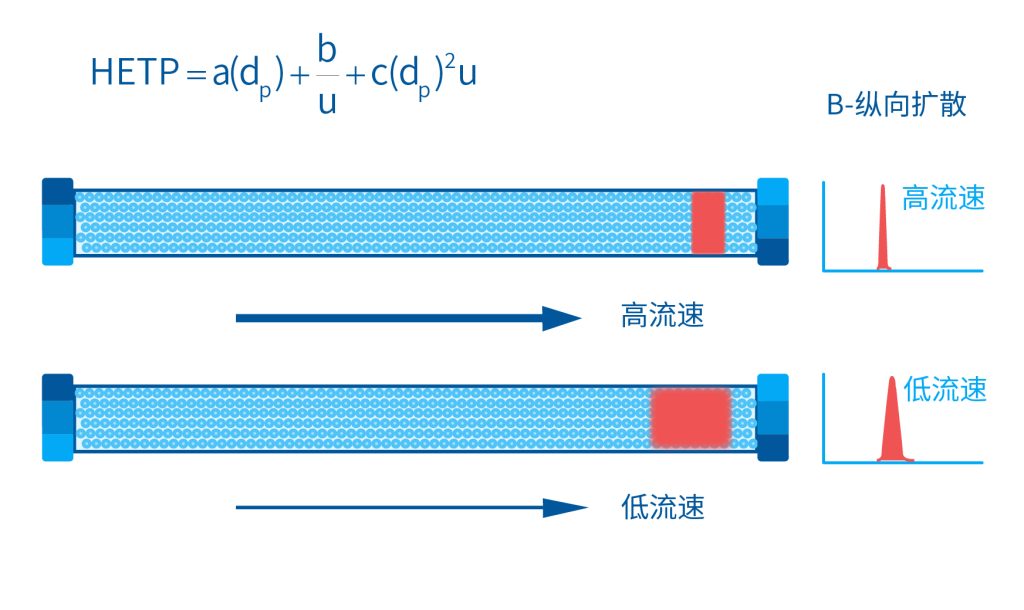
色谱柱分离原理 这期来介绍一下色谱的原理,前面铺垫了那么久,脱气机减少气体对于流动相的干扰,泵提供准确稳定的流速,进样系统让样品引入到流动相中,这些部件共同服务于色谱柱,这是色谱的灵魂,也有的老师说色谱柱的选择不仅仅是技术,而是一门艺术。 从我个人从业经历来讲,分析行业就是增加目标化合物信息维度的行业,比如色谱的出峰时间、峰高、紫外吸收、质荷比、MRM、高分辨质谱、淌度、XRD、红外、热失重、核磁等等,每一个都是增加待测物的信息维度,当有足够的信息维度,就可以更准确的对化合物进行更好的了解。 当年没干这行的时候,还停留在初中高中的化学知识,对物质的判断靠非常粗略的办法,其实就是信息维度低。 色谱说简单点就是一套分离系统,一个混合物里面,有多少种物质和多少量是不知道的,这时就利用不同物质在固定相和流动相之间的保留不同,进而经过色谱柱分离后,达到检测器的时间不同进行区分,也就是这个动画想表达的意思。 所以一张色谱图有两个最核心的信息,保留时间和信号强度,分别可以用来定性和定量,但这里有一个问题,一般认为进样后所有的物质都会从色谱柱流出,那么有没有些物质在固定相上面的保留特别强,你的色谱条件根本不能把它洗脱下来呢? 本人以前是做人体中毒的,按照标准方法处理完血清上机出结果,色谱图是很干净,但是做完所有样品冲洗色谱柱,提高有机相比例后,检测器会显示大量的物质从色谱柱流出,需要冲洗很长时间,相信您一定也会遇到类似的情况。 所以我在信息维度中多记录了一个压力,这是我个人认为液相色谱中最重要的一个参数,因为压力是两方便造成的,一个是系统本身,另外一个是色谱柱。 如果您每次进样,压力都能保证完美的一致,那么你的结果重现性一定非常高,说明你的系统和色谱柱的状态都非常好,您保留的异常都可以从压力上体现出来,建议您也更多的关注一下系统压力,特别是UPLC的用户。 请看样品进入流动相之后的变化,这个图简化的理想模型,是一滴红色的液体滴到有展开液的平面上,它自然会向周围扩散,这也就说明样品进入流动相之后就会发生扩散。 根据菲克定律,扩散的范围和浓度成正比,并且扩散会使浓度随时间改变,也就是随着时间这滴液体直径会变大,颜色会变浅,上面的色谱图是这滴液体的积分,这就是在色谱图上看到的峰,您可以简单的理解为,峰起点是这个液滴刚到达检测器的时刻,检测器出现了信号,这个液滴继续向前检测器信号变强达到最高值。 这个最高点是液滴浓度最高的地方,最后液滴离开检测器信号又回到基线。 这个峰和统计学上的正态分布非常像,峰高的地方是化合物分子,在统计学上分布最高的地方,两边降低是该分子分布概率降低,后面我们也会涉及到这种统计学的解释。 图中看到左侧液体的积分最高点,高于右侧的积分最高点,但左侧的液滴的直径是明显小于右边的,这是因为液相的检测器属于光学检测器,满足比尔定律,虽然右侧的l厚度长,但是左边的c浓度大,最终导致了这个结果。如果从理论上来讲,左右两个积分的面积应该是相同的。 这里引入一个概念谱带展宽,在被测物谱带到达检测器之前,将通过色谱系统的多个组成单元,这都使色谱谱带失真并加宽,这种现象称为谱带展宽,也就是我们刚才提到的扩散。 最上面的图红色代表样品进入流动相,因为进样针和定量环管路都有一定长度,所以样品是一段液体而不是球形的液滴,刚进入流动相时谱带展宽最小的,如果不考虑在定量环中的扩展,那么理论上这个样品的积分应该矩形的,因为各处的浓度是一致的。 中间第二幅图是样品不连接色谱柱直接到达检测器,这里谱带展宽的原因是系统流路造成的,也就是式中柱外效应的方差,造成了谱带展宽。 最下面的图是样品流经色谱柱到达检测器,谱带进一步展宽,总体方差是柱外方差和柱内方差之和。 在区分色谱柱的时候,柱效可能是用来描述色谱柱性能最常用的概念,以理论塔板数N来表示,您购买的色谱柱里都会附上色谱柱出厂的性能报告,就是用柱效作为指标。 图中只列出了柱效N的一种方程,因为柱效可以通过保留时间,不同位置的峰宽和相应的系数出现不同的方式,但理论上的结果都是一样的,从公式可以知道相同保留时间下,峰宽越小对柱效的提高越大,当然刚才提到的谱带展宽对峰宽是有影响的,所以要尽可能减少谱带展宽。 那我们要做些什么呢,对于不锈钢管路我们是没有办法的,可操作的是peek管路,在满足分析的情况下越短越好,所有peek管路的切割一定要平齐,peek管路一定伸到最里面再拧紧,这样减少死体积。 还有一点是管路的直径,Agilent不同内径的管路用不同颜色进行区分,您可以在满足系统使用的情况下,减少peek管的内径,比如色谱柱出口直接接质谱的话,用红色小内径的管路比较好。 下一个概念是保留因子k,是指样品组分停留在固定相中,相对其驻留在流动相中的时间之比,用保留时间除以峰不保留时间t0进行计算,也就是保留因子k越大,代表固定相对化合物的保留更强,出峰时间越晚。相反的保留因子k越小,代表流动相对化合物的保留更强,出峰时间越早,在常用的反向色谱中,出峰越早代表化合物的极性越强,反之出峰越晚代表化合物的极性越弱。 这里产生个问题,不保留时间t0该如何取得数据呢,一般是通过尿嘧啶作为测试样品得到的,因为尿嘧啶在C18柱上没保留,所以认为它的出峰时间就是死时间。 接下来如何描述两个峰位置的关系呢,一般用分离因子α来表示,分离因子是对两个色谱峰,时间或距离的最大测量值,如果α = 1,那么两个色谱峰具有相同的保留时间并共洗脱,这个时候在化合物信息维度不足情况下,很难判断峰的纯度。 这种情况在做复杂气相样品时候比较明显,由于复杂样品得到的峰非常多且密集,在FID检测器上的一个峰,在质谱信号中可能是多个峰的组成,在这张图中可以看到峰A和峰B之间的选择性,比峰B和峰C的选择性更好。 还有一个是分离度Rs,分离度代表色谱柱分离目标峰的能力,所以分离度越高,两峰之间越容易得到基线分离,从分离度方程中可以看出,分离度与柱效、选择性和保留值都相关,可以通过改善这些因素来提高分离度。 如果要对色谱峰进行定量,那么分离度最小应等于1,分离度要达到0.6,才能分辨两个等高峰之间的峰谷,如需建立耐用方法,理想分离度值一般应大于等于1.7,基线分离并确保最准确定量结果的分离度应为1.6。 既然我们知道了分离度公式,和塔板数N、选择因子α和保留因子k有关,我们看一下分别调整每个参数会发生什么样的变化。 首先是塔板数N,塔板数是物理性参数,由于该参数的平方根对分离度存在影响,实际中你很难调整一个色谱柱的塔板数N,除非您串联多跟色谱柱,但系统压力也会增加。如果颗粒非常小,塔板数对分离度将有显著的影响,通过改变塔板数峰的中心距不会改变,颗粒减小将使色谱峰更窄、效率更高,从而能提高分离度和灵敏度,这也是当前高端色谱都用小粒径的原因。 接下来是最常用的调整分离度的方式,选择因子α受到温度、洗脱液、流动相组成,和色谱柱的化学性质等因素的共同影响,所以调整流动相就可以非常方便的改变分离度,不过可能会造成出峰顺序的改变,这里也说明为什么要控制柱温,因为温度改变会使α受到影响,有可能改变色谱结果。 最后一个是保留因子,这个和选择因子差不多,而且两者彼此相关,我们能通过提高k来改善分离度,但是这样会导致分离结果的保留时间延长,灵敏度降低并且峰宽加宽,您可以根据您实际的需要来对这些参数进行调整,满足您的分析需要。 之前说过,峰宽可认为是被测分子的统计分布方差,峰宽与峰走过的距离成比例线性增加,峰宽和峰所走过的距离之间的关系是一个概念,被称为理论塔板高度H,这个概念来源于蒸馏理论,是柱效应的度量指标。 需要考虑几种谱带扩展的相关过程,踏板高度越小色谱柱中的塔板数N越多,如果能够弄清楚,柱内被测分子怎样与流动相和固定相相互作用,我们就能近一步理解发生的不同相关扩散的过程,以及这些过程如何影响了色谱性能,有三个同时发生的相关扩散过程。 图中的A项,被测分子被运送到颗粒表面和颗粒周围涡流扩散。 图中的B项,被测分子在大体积流动相中前后扩散的纵向扩散。 图中的C项,被测分子扩散进出色谱孔的质量传递。 他们共同组成了范第姆特方程式,描述了每一项与理论塔板高度之间的关系。 先来看一下涡流扩散A项,主要与填料的颗粒大小相关,其取值也由色谱床装填的好坏程度决定,如图中所示,最上面是小粒径的色谱柱,被测物分子从流动相向颗粒表面和颗粒周围转移的路径所需时间更少,峰宽更窄。 中间的图是化合物通过大粒径的色谱柱,明显可以看到化合物相比小粒径的色谱柱,要经过更长的距离和时间,而这种距离的增加会改变化合物分子的统计学分布,也就是让峰宽变宽。 最后一个图是填料的好坏,也包含填料直径的不均匀,这里画的比较夸张,但意思很明确,当填料质量非常好的时候,化合物分子的统计学分布更集中,峰宽更窄。当质量不佳的时候,化合物要经过非常曲折的路线和更多的时间,这就让化合物分子的统计学分布更分散,峰会变的更宽,涡流扩散与色谱柱线速度不相关,所以范第姆特方程中近乎一条直线。 第二项纵向扩散,与流动相和固定相上,被测物的扩散相关,随流动相速度的增加而减小,该项不依赖颗粒大小,它表示如果流动相以较慢的线速度移动,被测物分子将在色谱柱中停留更长的时间,因此在色谱柱中存在谱带纵向展宽的可能性更大,相反的如果流动相以更快的线速度移动,扩散时间更短,这样很少谱带展宽现象发生。 最后一个是质量传递,它与线速度和颗粒大小的平方相关,在化合物分子分布中,一些分子进入了固定相的小孔,其它分子则保留在移动的流动相中,直到遇到其它的固定相颗粒,随后发生相反的过程,被固定的分子脱离固定相,进一步向色谱床的下端移动。但是被测物分子进出小孔的程度不同,当分子返回到主体流动相时,每个被测分子经过的路径长度不同,这样导致被测物谱带展宽,谱带展宽的程度取决于流动相的速度。 另一方面取决于流速,分子进出固定相的小孔需要花费时间,当分子从一个固定相颗粒转移到下一个颗粒时,化合物谱带在色谱柱中移动将会变宽,如果流动相移动快速,那么在固定吸附分子与前面移动的分子之间将出现较大的距离,这说明为了整个被测物分子能够尽可能保持一起移动,应该采用比较慢的流速,当流动相的速度增加时,整个被测物分子将更加分散,结果谱带会扩展加大。 最后回到范第姆特方程,这个蓝色的线实际是由ABC三个虚线相加共同组成的,对于我们实际的分析,当然希望柱效N越高越好,这样可以得到更好的色谱结果。 根据公式变换理论塔板高度越低越好,对应图中蓝色线的最低点就是最优解,相对应的流速就是该粒径下的最优流速,所以对于我们常见的5μm粒径的色谱柱,这个最优流速是1 mL/min左右,这也就是为什么各种国标方法都用的流速在1 mL/min左右,在这个流速下,你用比较长的250mm的柱子压力也就200 bar左右,再高的压力您是很难用到的,不管最高是400 bar还是600 bar,除非是堵了。 不过您也要知道,当粒径变小的时候压力会增加,这时系统的最高耐压是有用的,这些内容会在UPLC原理中和大家探讨。 今天和大家聊了很多理论的知识,希望对您能有所帮助,有的同学会说,陶工你讲这么多理论对我们工作没用啊,让我们会操作就行了,最后这里想和大家分享一个我的亲身经历,当年科室要让我开发尿中锰元素的方法,之间的富集法很难用,而且不准确。 当时我们有一台原子吸收,一想买个灯就能干呗,有啥难的,前前后后忙了半个月,正好他们工程师来拜访和他聊起来这个事,他查了一下参数,告诉我别瞎忙了,这仪器锰的检出限测不出你样品中的最高浓度,当时直接给我震撼了,白忙了。 之前我们提到分析工作是增加化合物信息维度,而作为操作者也需要更高的维度看待分析问题,当年我当实验员的时候也只知道数据不准就是重做,一遍不行两遍,两遍不行四遍,而今天作为工程师回头去看当年遇到的问题就豁然开朗,因为可以从仪器的原理和结构设计上,快速判断结果异常是什么原因造成的,会提高非常多的效率,希望可以和大家一起进步。 联系 189 4564 8369 zitaikeji zitaikeji@gmail.com 地址 哈尔滨高新技术产业开发区科技创新城创新创业广场14号 链接 关于我们 联系我们 联系 189 4564 8369 zitaikeji zitaikeji@gmail.com 地址 哈尔滨高新技术产业开发区科技创新城创新创业广场14号 链接 关于我们 联系我们 色谱柱分离原理 这期来介绍一下色谱的原理,前面铺垫了那么久,脱气机减少气体对于流动相的干扰,泵提供准确稳定的流速,进样系统让样品引入到流动相中,这些部件共同服务于色谱柱,这是色谱的灵魂,也有的老师说色谱柱的选择不仅仅是技术,而是一门艺术。 从我个人从业经历来讲,分析行业就是增加目标化合物信息维度的行业,比如色谱的出峰时间、峰高、紫外吸收、质荷比、MRM、高分辨质谱、淌度、XRD、红外、热失重、核磁等等,每一个都是增加待测物的信息维度,当有足够的信息维度,就可以更准确的对化合物进行更好的了解。 当年没干这行的时候,还停留在初中高中的化学知识,对物质的判断靠非常粗略的办法,其实就是信息维度低。 色谱说简单点就是一套分离系统,一个混合物里面,有多少种物质和多少量是不知道的,这时就利用不同物质在固定相和流动相之间的保留不同,进而经过色谱柱分离后,达到检测器的时间不同进行区分,也就是这个动画想表达的意思。 所以一张色谱图有两个最核心的信息,保留时间和信号强度,分别可以用来定性和定量,但这里有一个问题,一般认为进样后所有的物质都会从色谱柱流出,那么有没有些物质在固定相上面的保留特别强,你的色谱条件根本不能把它洗脱下来呢? 本人以前是做人体中毒的,按照标准方法处理完血清上机出结果,色谱图是很干净,但是做完所有样品冲洗色谱柱,提高有机相比例后,检测器会显示大量的物质从色谱柱流出,需要冲洗很长时间,相信您一定也会遇到类似的情况。 所以我在信息维度中多记录了一个压力,这是我个人认为液相色谱中最重要的一个参数,因为压力是两方便造成的,一个是系统本身,另外一个是色谱柱。 如果您每次进样,压力都能保证完美的一致,那么你的结果重现性一定非常高,说明你的系统和色谱柱的状态都非常好,您保留的异常都可以从压力上体现出来,建议您也更多的关注一下系统压力,特别是UPLC的用户。 请看样品进入流动相之后的变化,这个图简化的理想模型,是一滴红色的液体滴到有展开液的平面上,它自然会向周围扩散,这也就说明样品进入流动相之后就会发生扩散。 根据菲克定律,扩散的范围和浓度成正比,并且扩散会使浓度随时间改变,也就是随着时间这滴液体直径会变大,颜色会变浅,上面的色谱图是这滴液体的积分,这就是在色谱图上看到的峰,您可以简单的理解为,峰起点是这个液滴刚到达检测器的时刻,检测器出现了信号,这个液滴继续向前检测器信号变强达到最高值。 这个最高点是液滴浓度最高的地方,最后液滴离开检测器信号又回到基线。 这个峰和统计学上的正态分布非常像,峰高的地方是化合物分子,在统计学上分布最高的地方,两边降低是该分子分布概率降低,后面我们也会涉及到这种统计学的解释。 图中看到左侧液体的积分最高点,高于右侧的积分最高点,但左侧的液滴的直径是明显小于右边的,这是因为液相的检测器属于光学检测器,满足比尔定律,虽然右侧的l厚度长,但是左边的c浓度大,最终导致了这个结果。如果从理论上来讲,左右两个积分的面积应该是相同的。 这里引入一个概念谱带展宽,在被测物谱带到达检测器之前,将通过色谱系统的多个组成单元,这都使色谱谱带失真并加宽,这种现象称为谱带展宽,也就是我们刚才提到的扩散。 最上面的图红色代表样品进入流动相,因为进样针和定量环管路都有一定长度,所以样品是一段液体而不是球形的液滴,刚进入流动相时谱带展宽最小的,如果不考虑在定量环中的扩展,那么理论上这个样品的积分应该矩形的,因为各处的浓度是一致的。 中间第二幅图是样品不连接色谱柱直接到达检测器,这里谱带展宽的原因是系统流路造成的,也就是式中柱外效应的方差,造成了谱带展宽。 最下面的图是样品流经色谱柱到达检测器,谱带进一步展宽,总体方差是柱外方差和柱内方差之和。 在区分色谱柱的时候,柱效可能是用来描述色谱柱性能最常用的概念,以理论塔板数N来表示,您购买的色谱柱里都会附上色谱柱出厂的性能报告,就是用柱效作为指标。 图中只列出了柱效N的一种方程,因为柱效可以通过保留时间,不同位置的峰宽和相应的系数出现不同的方式,但理论上的结果都是一样的,从公式可以知道相同保留时间下,峰宽越小对柱效的提高越大,当然刚才提到的谱带展宽对峰宽是有影响的,所以要尽可能减少谱带展宽。 那我们要做些什么呢,对于不锈钢管路我们是没有办法的,可操作的是peek管路,在满足分析的情况下越短越好,所有peek管路的切割一定要平齐,peek管路一定伸到最里面再拧紧,这样减少死体积。 还有一点是管路的直径,Agilent不同内径的管路用不同颜色进行区分,您可以在满足系统使用的情况下,减少peek管的内径,比如色谱柱出口直接接质谱的话,用红色小内径的管路比较好。 下一个概念是保留因子k,是指样品组分停留在固定相中,相对其驻留在流动相中的时间之比,用保留时间除以峰不保留时间t0进行计算,也就是保留因子k越大,代表固定相对化合物的保留更强,出峰时间越晚。相反的保留因子k越小,代表流动相对化合物的保留更强,出峰时间越早,在常用的反向色谱中,出峰越早代表化合物的极性越强,反之出峰越晚代表化合物的极性越弱。 这里产生个问题,不保留时间t0该如何取得数据呢,一般是通过尿嘧啶作为测试样品得到的,因为尿嘧啶在C18柱上没保留,所以认为它的出峰时间就是死时间。 接下来如何描述两个峰位置的关系呢,一般用分离因子α来表示,分离因子是对两个色谱峰,时间或距离的最大测量值,如果α = 1,那么两个色谱峰具有相同的保留时间并共洗脱,这个时候在化合物信息维度不足情况下,很难判断峰的纯度。 这种情况在做复杂气相样品时候比较明显,由于复杂样品得到的峰非常多且密集,在FID检测器上的一个峰,在质谱信号中可能是多个峰的组成,在这张图中可以看到峰A和峰B之间的选择性,比峰B和峰C的选择性更好。 还有一个是分离度Rs,分离度代表色谱柱分离目标峰的能力,所以分离度越高,两峰之间越容易得到基线分离,从分离度方程中可以看出,分离度与柱效、选择性和保留值都相关,可以通过改善这些因素来提高分离度。 如果要对色谱峰进行定量,那么分离度最小应等于1,分离度要达到0.6,才能分辨两个等高峰之间的峰谷,如需建立耐用方法,理想分离度值一般应大于等于1.7,基线分离并确保最准确定量结果的分离度应为1.6。 既然我们知道了分离度公式,和塔板数N、选择因子α和保留因子k有关,我们看一下分别调整每个参数会发生什么样的变化。 首先是塔板数N,塔板数是物理性参数,由于该参数的平方根对分离度存在影响,实际中你很难调整一个色谱柱的塔板数N,除非您串联多跟色谱柱,但系统压力也会增加。如果颗粒非常小,塔板数对分离度将有显著的影响,通过改变塔板数峰的中心距不会改变,颗粒减小将使色谱峰更窄、效率更高,从而能提高分离度和灵敏度,这也是当前高端色谱都用小粒径的原因。 接下来是最常用的调整分离度的方式,选择因子α受到温度、洗脱液、流动相组成,和色谱柱的化学性质等因素的共同影响,所以调整流动相就可以非常方便的改变分离度,不过可能会造成出峰顺序的改变,这里也说明为什么要控制柱温,因为温度改变会使α受到影响,有可能改变色谱结果。 最后一个是保留因子,这个和选择因子差不多,而且两者彼此相关,我们能通过提高k来改善分离度,但是这样会导致分离结果的保留时间延长,灵敏度降低并且峰宽加宽,您可以根据您实际的需要来对这些参数进行调整,满足您的分析需要。 之前说过,峰宽可认为是被测分子的统计分布方差,峰宽与峰走过的距离成比例线性增加,峰宽和峰所走过的距离之间的关系是一个概念,被称为理论塔板高度H,这个概念来源于蒸馏理论,是柱效应的度量指标。 需要考虑几种谱带扩展的相关过程,踏板高度越小色谱柱中的塔板数N越多,如果能够弄清楚,柱内被测分子怎样与流动相和固定相相互作用,我们就能近一步理解发生的不同相关扩散的过程,以及这些过程如何影响了色谱性能,有三个同时发生的相关扩散过程。 图中的A项,被测分子被运送到颗粒表面和颗粒周围涡流扩散。 图中的B项,被测分子在大体积流动相中前后扩散的纵向扩散。 图中的C项,被测分子扩散进出色谱孔的质量传递。 他们共同组成了范第姆特方程式,描述了每一项与理论塔板高度之间的关系。 先来看一下涡流扩散A项,主要与填料的颗粒大小相关,其取值也由色谱床装填的好坏程度决定,如图中所示,最上面是小粒径的色谱柱,被测物分子从流动相向颗粒表面和颗粒周围转移的路径所需时间更少,峰宽更窄。 中间的图是化合物通过大粒径的色谱柱,明显可以看到化合物相比小粒径的色谱柱,要经过更长的距离和时间,而这种距离的增加会改变化合物分子的统计学分布,也就是让峰宽变宽。 最后一个图是填料的好坏,也包含填料直径的不均匀,这里画的比较夸张,但意思很明确,当填料质量非常好的时候,化合物分子的统计学分布更集中,峰宽更窄。当质量不佳的时候,化合物要经过非常曲折的路线和更多的时间,这就让化合物分子的统计学分布更分散,峰会变的更宽,涡流扩散与色谱柱线速度不相关,所以范第姆特方程中近乎一条直线。 第二项纵向扩散,与流动相和固定相上,被测物的扩散相关,随流动相速度的增加而减小,该项不依赖颗粒大小,它表示如果流动相以较慢的线速度移动,被测物分子将在色谱柱中停留更长的时间,因此在色谱柱中存在谱带纵向展宽的可能性更大,相反的如果流动相以更快的线速度移动,扩散时间更短,这样很少谱带展宽现象发生。 最后一个是质量传递,它与线速度和颗粒大小的平方相关,在化合物分子分布中,一些分子进入了固定相的小孔,其它分子则保留在移动的流动相中,直到遇到其它的固定相颗粒,随后发生相反的过程,被固定的分子脱离固定相,进一步向色谱床的下端移动。但是被测物分子进出小孔的程度不同,当分子返回到主体流动相时,每个被测分子经过的路径长度不同,这样导致被测物谱带展宽,谱带展宽的程度取决于流动相的速度。 另一方面取决于流速,分子进出固定相的小孔需要花费时间,当分子从一个固定相颗粒转移到下一个颗粒时,化合物谱带在色谱柱中移动将会变宽,如果流动相移动快速,那么在固定吸附分子与前面移动的分子之间将出现较大的距离,这说明为了整个被测物分子能够尽可能保持一起移动,应该采用比较慢的流速,当流动相的速度增加时,整个被测物分子将更加分散,结果谱带会扩展加大。 最后回到范第姆特方程,这个蓝色的线实际是由ABC三个虚线相加共同组成的,对于我们实际的分析,当然希望柱效N越高越好,这样可以得到更好的色谱结果。 根据公式变换理论塔板高度越低越好,对应图中蓝色线的最低点就是最优解,相对应的流速就是该粒径下的最优流速,所以对于我们常见的5μm粒径的色谱柱,这个最优流速是1 mL/min左右,这也就是为什么各种国标方法都用的流速在1 mL/min左右,在这个流速下,你用比较长的250mm的柱子压力也就200 bar左右,再高的压力您是很难用到的,不管最高是400 bar还是600 bar,除非是堵了。 不过您也要知道,当粒径变小的时候压力会增加,这时系统的最高耐压是有用的,这些内容会在UPLC原理中和大家探讨。 今天和大家聊了很多理论的知识,希望对您能有所帮助,有的同学会说,陶工你讲这么多理论对我们工作没用啊,让我们会操作就行了,最后这里想和大家分享一个我的亲身经历,当年科室要让我开发尿中锰元素的方法,之间的富集法很难用,而且不准确。 当时我们有一台原子吸收,一想买个灯就能干呗,有啥难的,前前后后忙了半个月,正好他们工程师来拜访和他聊起来这个事,他查了一下参数,告诉我别瞎忙了,这仪器锰的检出限测不出你样品中的最高浓度,当时直接给我震撼了,白忙了。 之前我们提到分析工作是增加化合物信息维度,而作为操作者也需要更高的维度看待分析问题,当年我当实验员的时候也只知道数据不准就是重做,一遍不行两遍,两遍不行四遍,而今天作为工程师回头去看当年遇到的问题就豁然开朗,因为可以从仪器的原理和结构设计上,快速判断结果异常是什么原因造成的,会提高非常多的效率,希望可以和大家一起进步。 联系 189 4564 8369 zitaikeji zitaikeji@gmail.com